- Study objective: e.g. no more hypothesis testing.
- Study population: type or number, if it may restrict the objective; e.g. considerable sample size reduction; decrease in centres or geographical spread.
- Study design: e.g. follow-up type; passive versus patient diary.
Type IB category C.I.11.z
- Updates of RMPs not falling within the scope of type II variations (see above) are in principle type IB variations.
- Addition, modification or deletion of a safety concern (identified risks, potential risks, missing information) which has already been assessed and requested by the PRAC/CHMP in a previous procedure; i.e. the changes have already been formally assessed and agreed in principle as part of a previous procedure (e.g. assessment of signals, PSURs, variations, PAMs) by the PRAC/CHMP, although the agreement on the exact wording to be implemented in the RMP is still pending and further assessment is therefore required.
Note: In order for the implementation of pre-agreed RMP changes to be handled as a type IB variation, no additional data should be needed or submitted to support the proposes changes.
- Change to the final results due date i.e. the date for the provision of the final study report for category 1, 2 or 3 studies in the RMP and/or the Annex II, as relevant.
- Changes of a due date for protocol submission for an imposed study.
Note: Because no specific changes are identified by the variations classification guideline as falling by default into the type IB variation category, the above changes lead to such variation only when they constitute the reason for submitting the updated RMP. In case the MAH takes the opportunity to propose such changes with an RMP update undertaken for another reason (e.g. as part of a type II variation), these changes are accepted as minor and do not trigger additional variation scopes (please refer to Question 5 below).
Type IAIN category C.I.11 a)
- Implementation of changes to the conditions based on an exact wording agreed by PRAC/CHMP without any further changes, provided that no linguistic review of translations is required in case of simultaneous changes to the Annex II (i.e. deletion of information, changes to timelines are acceptable but not the implementation of new wording as such).
- Update of the RMP in response to a request following signal detection provided an exact wording agreed by PRAC/CHMP is implemented without further changes.
- Update of the RMP in response to a request following assessment of a protocol of a category 1,2 or 3 study provided an exact wording agreed by PRAC/CHMP is implemented without further changes.
Note: The changes to be implemented must already have been assessed by the Committee(s) in a previously concluded procedure; only the exact agreed wording is implemented, no additional changes are proposed and no further assessment is required.
However, it should be noted that it is rare that an exact wording is pre-agreed and therefore in most cases a type IB or type II variation will be required. Regardless, the MAH should always specify in the submission whether or not the proposed changes have already been assessed, and if so, as part of which procedure.
5. Which changes can be included in an RMP update without the need for an additional variation?
It is in principle acceptable to take the opportunity of a regulatory application (e.g. a type IB or type II variation) which warrants an update of the RMP to implement also:
- minor administrative changes to the RMP;
- template-related updates (e.g. from RMP template rev. 1 or rev. 2);
- updates of clinical / post-marketing data (e.g. exposure data and data coming from important clinical trials without impact on key safety information or final due dates);
- changes to category 4 studies listed in table III.4.4 (stated additional pharmacovigilance activities, also known as 'REC'= Recommendation) (only for RMPs using rev. 1 of the RMP template);
as long as the proposed changes are not affecting the summary of the safety concerns, the summary table of additional pharmacovigilance activities, the routine risk minimisation activities recommending specific clinical measures to address the risk or additional risk minimisation activities.
Further, in the event that relatively minor RMP changes are requested by PRAC/CHMP for implementation at the 'next regulatory opportunity', i.e. as part of the next application resulting in more substantial changes to the RMP (e.g. type IB variation, type II variation, line extension, renewal), these changes can be included as part of the next upcoming RMP update under a single scope i.e. without any need for an additional specific variation, unless there is a defined timeframe by when the update is requested and there is no other planned major RMP update in the same timeframe.
6. Can I group my RMP updates?
Each proposed 'major change' to the RMP triggers in principle its own type II variation scope.It should be noted that one specific type II variation is required for each scope even when submitted together with other major changes as part of a grouped variation application. The same rules apply to the grouping of major RMP changes as to the grouping of any other (non)clinical type II variations:
- changes meaningful to be reviewed simultaneously can be grouped;
- non-clinical and clinical safety changes are not accepted as part of the same grouping;
- and grouping should not delay the implementation of important changes (for instance a proposed extension of indication should not be grouped with safety variations).
With regard to multiple 'minor changes' which can be assessed as type IB variations if submitted on their own, these do not require a grouped application; instead it is acceptable to include these minor changes as part of one single type IB variation or type II variation without the triggering of additional type IB variation scopes i.e. any need for additional variations (see also Question 5 above).
The following cases are meant to illustrate how these rules would be applied for RMP updates:
Addition of a new Adverse Drug Reaction and a relevant warning to the SmPC via a type II variation C.I.4 with consequential update of the list of important identified risks in the RMP and submission of a final study report for a category 3 study in the RMP via a type II variation C.I.13 with consequential updates of the RMP (i.e. removal of the study from the Pharmacovigilance Plan). This can be submitted as a grouped application of 2 type II variations.
Submission of a final study report for a category 3 study via a type II variation C.I.13 with consequential updates of the RMP, and:
- Deletion of the category 3 study in the RMP – no need for separate variation since related to the main application;
- Addition of a safety concern in the RMP following a request from PRAC as part of a PSUR assessment – 1 (grouped) Type II category C.I.11.b if additional data are submitted and/or further significant assessment is required; no need for a separate variation otherwise as the change is implemented as part of a type II variation affecting the RMP;
- Changes to the due date for the provision of the final study report for a category 3 study in the RMP – no need for a separate variation as the change is implemented as part of a type II variation affecting the RMP;
- Update of the RMP with significant changes of the clinical trial exposure – can be implemented within the variation without the need for an additional variation.
Changes to the due date for the provision of the final study report for two category 3 studies in the RMP.
This can be submitted as a single type IB variation under category C.I.11.z.
On the other hand, a grouped application is generally not acceptable if it creates the risk of postponing the implementation of important safety information in the RMP:
- In case a type II variation is submitted under category C.I.6 (Extension of Indication), the RMP version submitted as part of this application should include changes that are consequential to the new data provided and the new proposed indication, and it can also include changes that have been previously assessed and agreed.As the procedure for an extension of indication application may take some months to finalise, other non-related changes that require assessment should not be included and/or grouped with an extension of indication application (e.g. the implementation of safety information should not be delayed).
7. How should I handle parallel RMP submissions?
There is only one approved RMP at any time for a medicinal product. Consequently, any time an updated RMP is approved as part of a procedure (e.g. variation, renewal, PSUR), this RMP becomes the approved RMP of the product, and any previous version becomes obsolete. Therefore, MAHs should carefully consider the planning of RMP submission, to make sure that the approved RMP always contains the most up-to-date information on the pharmacovigilance planning and risk minimisation measures.
Given the content-based requirements for RMP submission, it is expected that there will be only few procedures where an RMP update should be included. The MAH should consider whether an RMP is really required with the procedure that is in preparation for submission. Early discussion with the regulators should facilitate the submission, to avoid unnecessary RMP submissions and assessment; parallel procedures warranting RMP updates should be avoided as much as possible.
MAHs are strongly encouraged to streamline RMP amendments and submissions, in co-operation with the EMA for the centrally authorised products, in order to facilitate RMP assessments throughout the product life-cycle.
There are two alternative approaches to the handling of different RMP versions for which the assessment is overlapping, and the MAH should choose the option that facilitates the assessment taking into account anticipated timelines for the finalisation of the procedures.
Whenever separate applications affecting the RMP are submitted in parallel, in order to facilitate the review, it is generally agreed that the MAH initially and whenever appropriate during the procedure submits one joint draft RMP file as a 'working document'. This single RMP document should include all data consequential to the concerned procedures running in parallel . To facilitate the assessment, the proposed RMP changes should be marked (e.g. with different colour code), to differentiate changes specific to each procedure (example: new safety concerns derived from an extension of indication in a new population should be marked differently from the changes in the Pharmacovigilance Plan consequential to (early) termination of a study and initiation of another one as a consequence of the limited safety data gathered in the ended study).
If the parallel applications reach the finalisation stage at the same time, the consolidated RMP version will be adopted by the relevant Committee and will become the approved version of the RMP.
If the applications do not reach the finalisation-stage at the same time, at the time of the first opinion for the parallel procedures, the MAH will need to provide for review and approval a final RMP version including only the agreed changes related to the scope of the variation application for which the CHMP is about to adopt an opinion. The joint RMP 'working document' will continue to be used in the context of the remaining ongoing procedure(s).
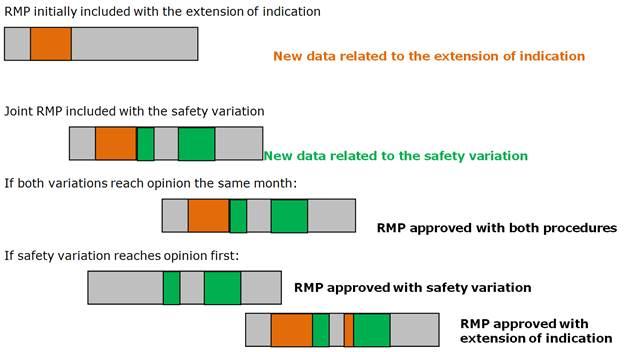
Example: A safety variation is triggered whilst an extension of indication procedure is ongoing, both requiring significant changes in the RMP (new safety concern in the new indication; another safety concern and a new imposed PASS in the safety variation). The RMP for the safety variation can be built upon the RMP document submitted with the extension of indication.
Option A: A joint RMP document including changes relevant to both procedures could be submitted with both the responses to the RSI in the extension procedure, and with the initial submission for the safety variation:
- If both procedures reach the Opinion stage at the same time, than the joint RMP will be adopted and become the approved RMP.
- If however the extension of indication requires a second RSI, and is most likely to be finalised after the parallel safety variation, the MAH will then have to submit before the opinion for the safety variation an RMP including only the safety concern and the new study related to the safety variation data. This version of the RMP will be checked for consistency and approved with the safety variation opinion. The updated joint RMP 'working document', including the changes consequential to the responses to the second RSI for the extension of indication will continue to be assessed within the extension of indication procedure. This joint RMP will be considered the approved RMP once the extension of indication variation reaches opinion.
A graphical representation for option A is included below:
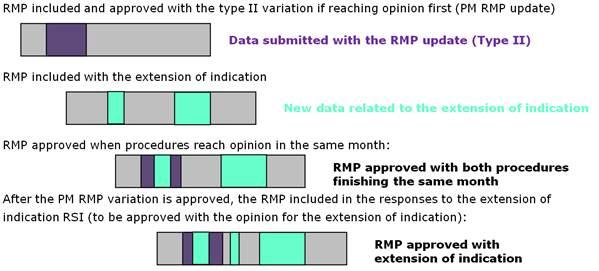
Option B: Alternatively, it might be more appropriate when parallel procedures will follow very different assessment timetables to opt for an approach similar to the handling of parallel procedures with product information changes; the RMP submitted with each procedure should only include the changes related to that procedure:
- an updated version of the RMP is submitted as a type II variation to reflect the changes in the safety profile derived from post-marketing reporting. This RMP version should include only the changes related to the RMP update.
- subsequently or at the same time, another RMP update is submitted as part of a type II variation for the extension of indication. For this application, the RMP version only includes the changes that are consequential to the extension of indication (i.e. not the changes related to the safety variation.
If both procedures conclude at the same time, the MAH is expected to merge the two RMP documents for approval by the Opinion time.
If the RMP update variation is approved before the extension of indication procedure, the RMP submitted will be adopted with the relevant changes and the MAH can submit a consolidated RMP version as part of the MAH's responses to an RSI for the extension of indication. This RMP version includes then the changes approved as part of the recently finalised safety variation (as clean text) as well as the changes related to the extension of indication (with track changes).
The option B can be illustrated as follows:
Regardless of the approach chosen, the MAH should always provide a clear description of the scope(s) of the submission in the cover letter and the changes implemented in the RMP including references to related (previous/parallel) regulatory procedure(s) (see also Question 8 below).
8. How shall I present my RMP update?
Guidance on the format and content of the RMP as outlined in GVP module V and RMP template has been made available in the Pharmacovigilance section of the Agency's website. The submitted RMP should follow the RMP template and guidance.
The RMP should be provided in CTD section 1.8.2. RMP versions submitted for assessment should be version controlled and dated. All parts and modules of the RMP should be submitted in one single PDF-file so that a complete RMP is provided to the Agency.
Only clean versions of documents in PDF format should be managed within the eCTD lifecycle. However, due to the fact that additional formats are required to facilitate the assessment i.e. 'tracked changes' versions for SmPCs, RMPs or other documents as specified by the agency, these should be provided in Word format in the separate folder 'XXXX-working documents'. Further details in this regard can be found in section 2.9.9 of the Harmonised Guidance for eCTD Submissions in the EU. It is generally not necessary to include the annexes as part of the RMP 'working document' unless any of the annexes are actually revised. If no tracked changes version can be compiled (e.g. due to template transition when the tracked changes would be significant throughout the document), a 'clean' Word version file of the RMP should still be submitted in the 'XXXX-working documents' folder; this will facilitate the preparation of the RMP Summary to be published on the Agency website.
In general, any submitted version of the RMP should be based on the latest approved version (i.e. the latest version agreed by CHMP). However, sometimes it may be more appropriate to base the next version to be submitted on the latest RMP 'working document' version, especially when several procedures affecting the RMP are ongoing in parallel (see Question 7 above).
Regardless, the submitted RMP version should be seen as a draft, until approved. Details of the RMP approval status should be provided in the Module I of the document. The revised RMP should always get a new version number every time an updated RMP version is submitted for assessment (see recommendations on document versioning in the Guidance on the format of the risk management plan (RMP) in the EU – in integrated format).
When relevant, a discussion of the proposed RMP changes should be included in the (non-) clinical overview (addendum). It should be noted that the provision of a (non-)clinical overview (addendum) is mandatory as part of a (non-)clinical type II variation application which includes a revised RMP regardless of the fact that there may be no impact on the product information. In this case the (non-) clinical overview (addendum) should discuss and justify the proposed RMP changes. On the other hand, a (non-) clinical overview (addendum) is never required as part of type IA and type IB variation applications.
In the EU application form (AF) and for (non-)clinical variations, the “Present/Proposed” table will in general only reflect proposed changes to the EN Annexes (SmPC, Annex II, labelling and Package Leaflet). It is not foreseen that the updates to the RMP are reflected in the AF in detail unless quite limited in scope. Instead, when comprehensive changes to the RMP are proposed, it is recommended to provide a comparative table of the RMP (latest agreed version vs. proposed version), summarising – for all individual RMP parts and modules – the main updates. For example, all changes linked to the implementation of a new template can be summarised as 'new RMP template'. Such comparative table should be provided as an annex to the AF.
9. Can I submit a version of the RMP after the Opinion to reflect the last minute changes made during the CHMP?
As a matter of principle the day of the CHMP Opinion/EMA Notification is the last opportunity for the MAH to provide an updated version of the RMP (in word format) for agreement. The same RMP version with the same version number – without any additional changes - can thereafter be submitted as part of a formal eCTD closing sequence post-opinion. However, if additional changes to the RMP are identified post-opinion after receipt of the document, an updated RMP version with a new version number should be provided for review as part of a type IB variation under category C.I.11.z.
The same principles apply also in situations when there are different RMP versions undergoing assessment in parallel and concluding the same month (see also Question 7 above). MAHs are requested to provide the final consolidated RMP version (in word format) before the date of the CHMP Opinion/EMA Notification.
10. Is the PRAC Rapporteur involved in the assessment of RMP updates?
The PRAC Rapporteur will be involved in the assessment of all variations that include an updated RMP. For type IB variation including RMP, PRAC Rapporteur will be in the lead of the assessment. For type II variations, the CHMP or PRAC may take the lead during the assessment depending on the composition of the data provided, and this will be decided on a case-by-case basis at the time of the EMA validation.
Similarly, on a case-by-case basis, the PRAC Rapporteur may also later become involved in the assessment of an application if requested by the CHMP during the procedure.
At the time of validation the Agency will inform the MAH of the involvement of the PRAC Rapporteur through the assessment timetable which will refer to the relevant assessment reports expected from the PRAC Rapporteur, as appropriate.
11. How long after the European Commission decision should Annex 1 of the RMP be submitted to EudraVigilance? Rev. Jun 2023
The maintenance of the Agency’s database for RMP Annex I files (‘Annex I tool’) has been suspended. Following this decision, as of 4th December 2020 the Marketing Authorisation Holders for centrally authorised products will no longer be required to create and submit RMP Annex I (.xml) files to the EMA.
Development and integration of a new database for structured electronic representation of the EU risk management plan to replace Annex I tool is pending the Agency’s digitalisation strategy. Any announcements about the system replacing the RMP Annex I database, reinitiating the requirements for the submissions, or otherwise instructions about the submissions of RMP in a structured electronic format will be provided (i.e. via the Agency website or directly to MAHs) as appropriate, in due time.
12. How and to whom shall I submit my RMP application?
As explained in the hereby questions and answers on RMP, the RMP update can be submitted either as part of a procedure driven by another main change defining the procedure classification (e.g. extension of indication, new formulation, etc.) or as a stand-alone variation exclusively including the RMP. In the latter, the variation can be either a type II, type IB or type IA, see question ‘Which variation classification will apply for my RMP updates?’ for further guidance.
Irrespective whether the RMP update is consequential to another change or a stand-alone update, the RMP document follows the eCTD life-cycle management and should be provided in Module 1.8.2 of the eCTD structure. Submission of the RMP should be made according to the framework of the procedure to which it belongs to and should follow the requirements and technical process for this procedure. More information is available on ‘Submitting a post-authorisation application’.
The use of the eSubmission Gateway or Web Client is mandatory for all electronic Common Technical Document (eCTD) submissions through the centralised procedure. The European Medicines Agency (EMA) no longer accepts submissions on CD or DVD. This applies to all applications for human medicines.
13. What templates should I use for the RMP submission?
Depending on the application submission date, either the Guidance on format of the risk-management plan in the European Union – in integrated format (Rev. 1) or the Revision 2 version of the Guidance on format of the risk-management plan in the European Union should be used including for generics. The Rev. 2 version is also applicable to generics as it includes specific guidance to generics. The transitional arrangements for the RMP submission are presented in the table below.
Acceptable template revisions to be used for RMP submissions:
RMPs submitted using Rev. 1 of the template instead of Rev.2 will not be rejected at validation of the submission, but will automatically trigger an additional step of assessment and an outstanding issue; applicants and MAHs will be required to update the RMP using the Rev.2 of the template and submit it with the responses to the RSI.
14. When and how will the full RMP be published on the EMA website? Rev. Dec 2023
All post-authorisation RMP updates assessed and approved in procedures concluding on or after 20 October 2023 will trigger the publication of the full RMP (body and Annexes 4 & 6).
For RMPs submitted for evaluation with type IB and IAINvariations, the MAH is asked to include the redacted version for publication (clean and tracked, redacting personal data and commercial confidential information) with the working documents in the variation eCTD sequence, together with the signed RMP Publication Declaration. It is recommended that all necessary changes are implemented via anonymisation and deletion directly in the RMP submitted for evaluation, rather than by redaction in the document for publication.
For RMPs submitted for evaluation in all other types of post-authorisation procedures, post-opinion/recommendation the MAH will be asked to extract the redacted RMP body and Annexes 4 & 6 (as applicable, redacting personal data and commercial confidential information) as one stand-alone PDF document and send it via EudraLink to the EMA, together with a RMP file that can show the content that is proposed for redaction, and the signed Template: Declaration for the risk management plan (RMP) publication.
The redacted RMP PDF will be published on the EMA website at the time of the EPAR update, on the product’s page (EPAR summary landing page).
15. How should I compile the list of safety concerns in the RMP for generic products when the originator products have an RMP?
When the MAH / Applicant for a generic medicinal product submits an RMP for assessment, the safety concerns should be aligned to those of the originator product that are available either from the originator’s approved RMP or from the list of safety concerns of the substance published on the CMDh website. Any divergence introduced in the RMP of the generic product (e.g. new safety concerns) should be thoroughly justified based on either differences in products’ characteristics (e.g. excipients), or on compelling data generated with this generic product that would warrant a difference in the list of safety concerns in the RMP (e.g. clinical trial or post-marketing epidemiological study data). This justification should be detailed in Module SVII of Part II of the RMP.
This guidance also applies on other types of marketing authorisations with similar RMP requirements: hybrid products and fixed combination products with no new active substance.